英文原题:Theoretical Perspectives of Precision Chemistry
通讯作者:李震宇,中国科学技术大学;杨金龙,中国科学技术大学
作者:Huan Ma (马欢), Jie Liu (刘杰), Zhenyu Li* (李震宇), Jinlong Yang* (杨金龙)
研究背景:
精准化学的核心目标是以100%的产率和选择性合成目标化合物,实现对化学反应和材料性能的精确预测与控制。实现这一目标,离不开对化学体系中电子和分子相互作用的深入理解。尽管实验表征手段日益强大,理论建模在机制探索和化学理解中仍占据主导地位。通过求解量子力学和统计力学方程,理论计算化学可以为分子和材料体系提供定量描述和预测模型,从而指导实验优化分子结构、反应路径和工艺参数。电子结构理论是理论化学的基础,电子结构计算精度直接影响统计和动力学性质预测的可靠性。然而,传统方法在精度与效率之间难以两全:平均场近似(如密度泛函理论)计算成本低,但在强关联体系中往往失效;高精度波函数方法(如完全活性空间组态相互作用)虽能系统描述电子关联,但计算量随体系规模呈指数增长。如何突破这一困境,是精准化学面临的关键挑战。
文章亮点:
近日,中国科学技术大学杨金龙、李震宇教授团队在Precision Chemistry上发表了题为“Theoretical Perspectives of Precision Chemistry”的前瞻性展望论文。文章系统总结了人工智能(AI)与量子计算(QC)在电子结构理论中的最新进展,并提出了构建“通用量子化学模型”的宏伟愿景,为精准化学的发展奠定了新的计算基础。

图1. 人工智能与量子计算驱动的量子化学近期进展
AI与量子计算赋能电子结构计算:文章指出,人工智能与量子计算正从三个关键层面重塑量子化学计算。首先,在波函数表示方面,神经网络量子态能够高效表示多电子波函数,结合变分量子本征求解器等量子算法,为复杂体系的电子结构描述提供了新途径。其次,在精准性质预测方面,AI与量子计算不仅可计算基态能量,还能拓展至激发态、原子力、光谱性质等,支持高精度分子动力学模拟。最后,在多尺度模拟方面,通过嵌入策略将高精度方法应用于关键活性区域,而环境部分采用低精度描述,从而高效处理生物大分子、材料界面等大体系。这三个层面的突破共同推动了精准化学计算能力的跨越式发展。
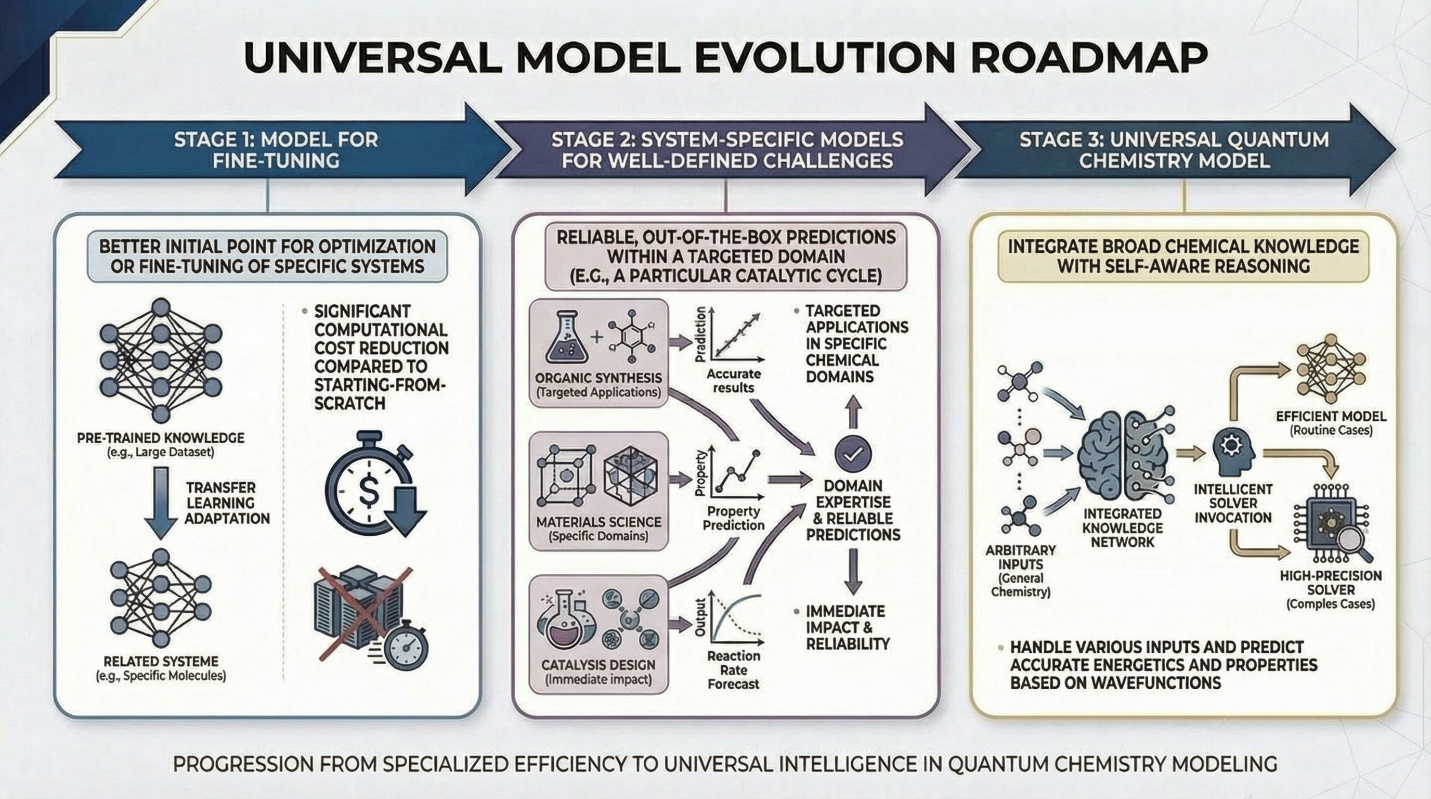
图2. 构建通用量子化学模型的发展路线图
迈向通用量子化学模型:受大语言模型成功启发,文章提出了“通用量子化学模型”的概念:一个预训练的、可迁移的计算框架,能够对从单参考分子到强关联固体等广泛化学体系,提供高精度电子结构和性质预测。研究团队展示了初步证据:基于Transformer的基态神经网络量子态可在不同哈密顿量间迁移;预训练模型经轻量微调即可适应新体系;Orbformer模型在2.2万分子结构上预训练后,能准确描述化学键断裂等强关联过程。
实现这一目标仍面临挑战:需设计融合原子结构信息的高表达力波函数架构,构建涵盖多样化学体系的高保真数据库。研究团队提出了三步走路线图:第一阶段,通用模型作为优化起点,降低计算成本;第二阶段,针对特定化学问题(如催化循环)发展领域模型;第三阶段,最终实现具备广泛化学知识与推理能力的通用量子化学模型。
总结/展望:
杨金龙、李震宇教授团队系统总结了AI与量子计算在量子化学中的前沿进展,指出二者正在为电子结构理论建立新的计算基础。神经网络量子态提供了经典可扩展的高表达性波函数,量子计算则开辟了可能实现量子优势的新硬件路径。通过生成高精度波函数和全面性质数据,这些方法有望从根本上重构化学发现的流程。
展望未来,通用量子化学模型将实现从传统模拟器到“化学智能”的跃迁,能够即时、可靠地推断复杂关联效应。理想情况下,量子计算生成的高精度数据用于训练AI模型,AI模型反过来提升量子化学计算的效率与广度,形成自进化的闭环生态,加速精准化学的发展。
Cite this: Huan Ma, Jie Liu, Zhenyu Li*, Jinlong Yang*. Theoretical Perspectives of Precision Chemistry. Precision Chemistry 2026. https://doi.org/10.1021/prechem.5c00382.