英文原题:Efficient Synthesis of Enlicitide Chloride Through Convergent Solution Phase and Hybrid Solution-Solid Phase Strategies
通讯作者:金康,山东大学药学院;方浩,山东大学药学院;宋伟国,山东道合药业有限公司
作者:Xuting Zhao(赵许亭), Xinqi Li(李鑫琦), Shuxian Zhang(张淑娴), Weiguo Song*(宋伟国), Hao Fang*(方浩), Kang Jin*(金康)
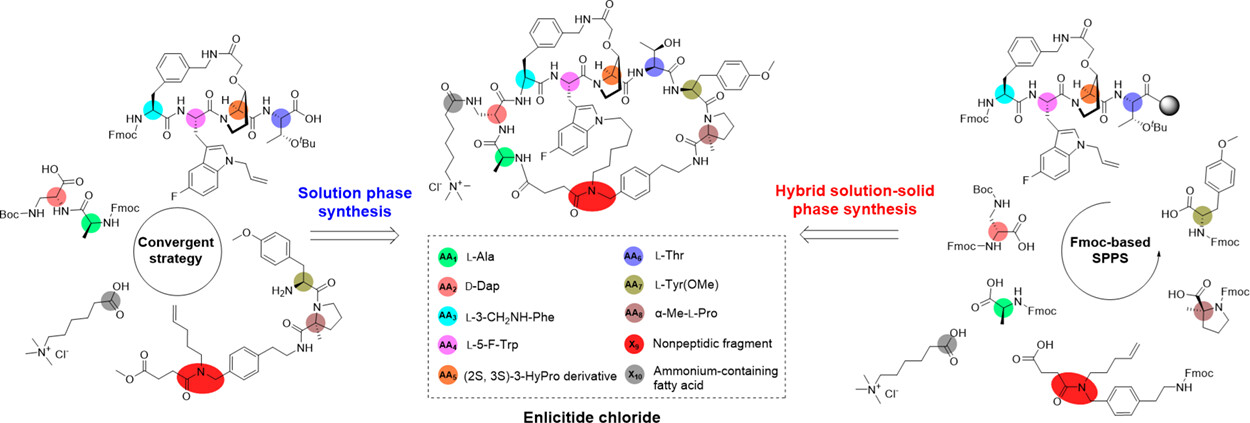
研究背景:
动脉粥样硬化性心血管疾病(ASCVD)是全球范围内导致死亡和发病的首要原因,涵盖冠心病、心肌梗死、心力衰竭、中风等多种病症,每年新增病例达数千万之多。高血脂是诱发该类疾病的主要危险因素,其中低密度脂蛋白胆固醇(LDL - C)水平升高尤为关键。目前,他汀类药物是临床首选的降脂药物,但长期服用可能引发肌病、肝功能异常、出血性中风及 2 型糖尿病等副作用,且约半数患者服用后仍无法将 LDL - C 控制在健康水平,其疗效和安全性存在明显局限。
前蛋白转化酶枯草溶菌素 9(PCSK9)在脂质代谢中扮演关键角色,它通过与低密度脂蛋白受体(LDLR)结合并促使其进入溶酶体降解,进而升高血清 LDL - C 水平,成为新型降脂药物的核心靶点。目前已开发的 PCSK9 抑制剂包括小分子化合物、蛋白疫苗、小干扰 RNA、单克隆抗体及多肽等,其中多肽类抑制剂凭借高效、低毒、靶向性强及成药性可优化等优势,成为降脂药物研发的热点方向。
Enlicitide(又称 MK - 0616)作为一种口服生物利用的大环肽类 PCSK9 抑制剂,目前正处于 III 期临床试验阶段,在 I、II 期临床试验中展现出优异的降脂效果和安全性,有望成为首个获批的口服大环肽 PCSK9 抑制剂。但该药物结构极具复杂性,是一种含铵侧链的三环肽,包含天然氨基酸、非蛋白原氨基酸及非肽片段等多种特殊结构单元,其合成所需的中间体难以获取,合成难度极大。尽管已有研究报道了液相片段缩合策略,但现有方法在正交保护基选择、中间体纯化及衍生物小规模合成等方面仍有很大改进空间,开发更高效、灵活的合成路线对Enlicitide的进一步研发至关重要。
内容介绍:
近日,山东大学药学院金康和方浩教授团队联合山东道合药业有限公司在 Precision Chemistry 上发表了氯化Enlicitide的高效合成研究。该研究制备了正交保护的非天然氨基酸片段,开发了片段缩合液相(4+3+2+1)合成法与液 - 固相杂交合成法,解决了复杂三环结构合成及中间体纯化难题。
团队成功合成了多种关键非天然氨基酸片段,且均带有完全正交的保护基,适配两种合成路径且无错脱保护现象。其中(2S, 3S)-3 - 羟脯氨酸衍生物 7 从商用原料出发,经甲酯化、醚化、保护基操控等多步反应制备而成,最终产物无需纯化即可直接用于下一步反应。特别地,特殊片段 14 的合成经亲核取代、肟化、氢化多步反应,三步总收率达 40%,最终经酰胺化和保护基转换后以高收率获得,完美适配 Fmoc 固相肽合成需求。
非天然氨基酸 17 以 Fmoc - L - Trp (5 - F) - OH 为原料,经脱保护、 Boc 安装和烷基化三步反应,收率达 71%。含铵脂肪酸 24 通过 TEMPO/NaClO/NaClO₂氧化和三甲胺取代两步反应合成,总收率 75%;二肽 21 的制备过程中,烯丙基化和偶联反应收率分别高达 97% 和 98%,这些高收率的片段合成工艺为整体合成路线的高效性提供了坚实保障。
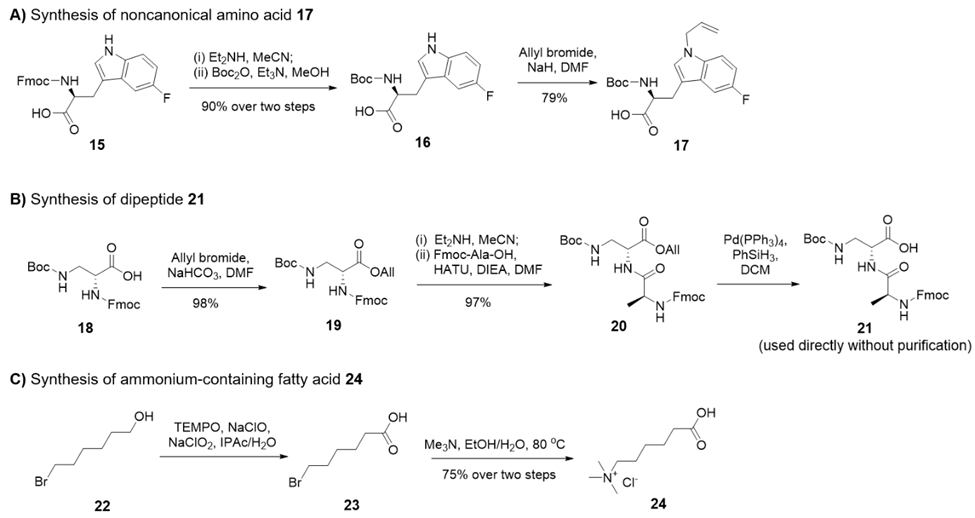
图1. 非天然氨基酸 17、二肽 21 及含铵脂肪酸 24 的合成
线性片段 29 的合成如图 2A 所示:化合物 13 脱 Boc 保护基后,与 Fmoc-α- 甲基 - L - 脯氨酸缩合得中间体 26;脱 Fmoc 保护基后再与 Fmoc-L - 酪氨酸(甲酯)-OH偶联制得骨架 28;脱除 N 端 Fmoc 保护基,即得片段 29,无需纯化可直接用于后续偶联。环状片段 39 的合成如图 2B 所示:片段 17 与 7 缩合生成二肽 30,经 30% 三氟乙酸 / 二氯甲烷脱 Boc 保护基得化合物 31;31 与 Fmoc-L - 苯丙氨酸(3 - 亚甲基氨基 - Boc)-OH酰胺化,两步总收率 95%,制得三肽 33。经脱烯丙酯保护基和脱 Boc 保护基得化合物 34,在 HATU/DIEA 作用下环化,三步总收率 87%,获环肽 35。用氢氧化锂脱除 35 的 Fmoc 保护基与甲酯基,重装 Fmoc 保护基得 36;36 与预先经烯丙基化、脱 Fmoc 保护基制得的 L - 苏氨酸(叔丁酯)- 烯丙酯偶联,生成环状四肽 38。脱除 38 的烯丙基,两步总收率 83%,得环状片段 39,该片段可满足液相合成与 Fmoc 固相肽合成(SPPS)需求。
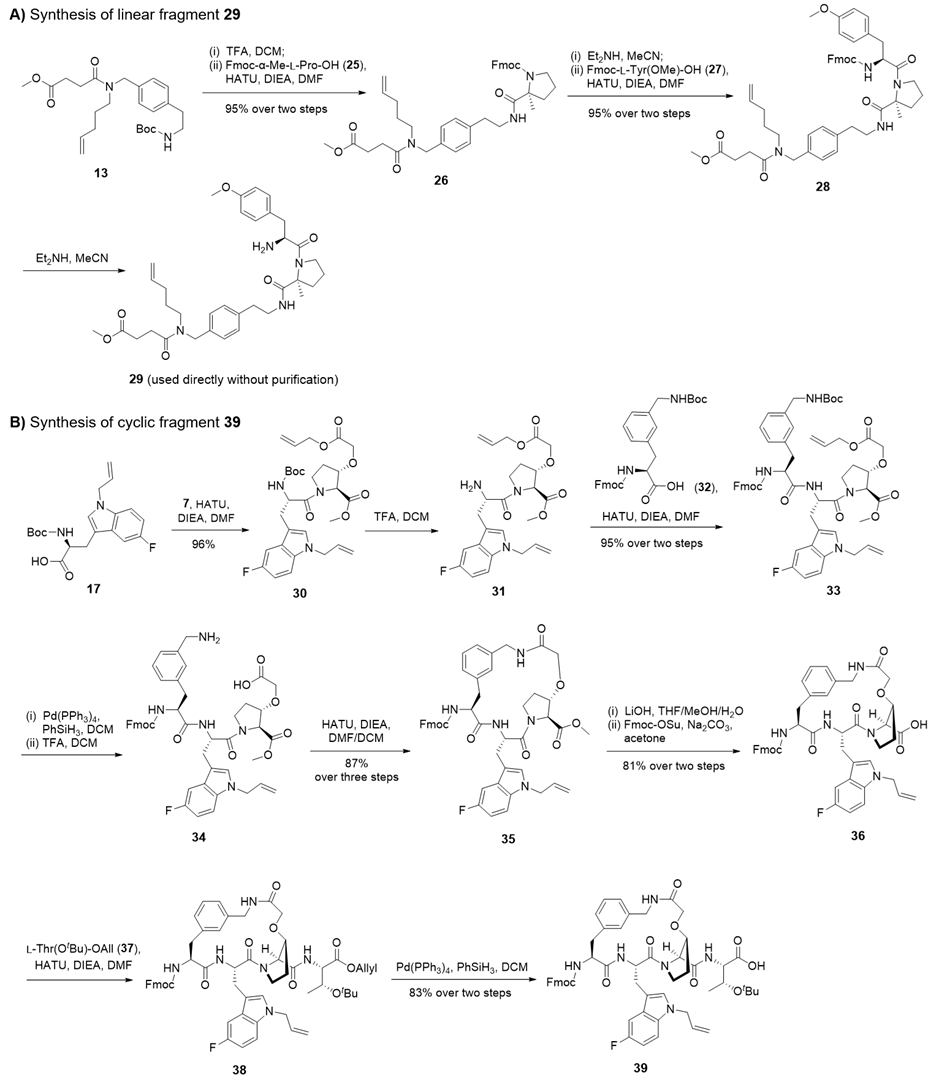
图2. 线性片段 29 与环状片段 39 的合成
片段缩合液相(4+3+2+1)合成法通过分步缩合不同片段,关键步骤收率表现优异。该路线先将环状片段 39 与线性片段 29 偶联得到肽 40,脱保护后再与二肽 21 缩合,两步总收率达 95%,成功构建出enlicitide核心全线性前体 41。在环化步骤中,团队借鉴前人经验优先进行丙氨酸(AA1)与非肽片段(X9)的分子内酰胺化,规避了副反应,HATU 介导的大环内酰胺化收率达 59%。随后经催化剂Zhan 1B 催化的关环复分解反应顺利形成三环骨架,收率高达 81%。最终,三环肽经催化氢化、脱保护后与含铵脂肪酸 24 偶联,四步总收率达 42%,经 HPLC 纯化后成功获得氯化enlicitide,整体路线稳定性与可靠性突出,适合工业化大规模生产。
液 - 固相杂交合成法创新性地解决了固相合成中环肽片段位阻大的难题,将环状片段 39 固定在 2 - 氯三苯甲基氯树脂上,通过标准 Fmoc 固相肽合成技术依次延伸 Fmoc - D - Dap (Boc) - OH、Fmoc - L - Ala - OH 等氨基酸链。针对固相合成中 Fmoc 脱保护反应速率较慢的问题,团队通过延长反应时间至约 1 小时,确保脱保护完全。肽链组装完成后,采用三氟乙醇 / 二氯甲烷 / 乙酸的温和酸性条件切割树脂,最大程度保留了肽段的完整性。该路径中,14 步反应获得双环肽 42 的收率为 38%,后续经关环复分解、氢化、脱保护、侧链安装等五步反应,收率达 34%,从树脂开始经 19 步反应实现总收率 13%。该方法有效避免了肽段差向异构化,简化了每步偶联后的纯化流程,大幅提升了合成效率,尤其适合中小规模的衍生物合成。

图3. 基于液 - 固相肽合成联合策略的enlicitide全合成
总结/展望:
综上,本研究以商品化原料为起始物,便捷制备了一系列含完全正交保护基的合成片段,这些片段可有效应用于后续肽合成,且无错脱保护现象发生。在此基础上,针对结构合成极具挑战性的enlicitide,开发出两种高效且可规模化的全合成方法,二者各具优势。具体而言,片段缩合策略的偶联效率优异(涵盖目标片段的液相合成与全肽序列的组装),可实现enlicitide的规模化生产,为其工业化制造提供全新且有价值的方案。另一种液 - 固相杂交合成策略,通过合理运用固相肽合成技术(SPPS),显著提升合成效率并节省时间成本,同时有效规避了潜在的肽段差向异构化问题,以及每步偶联反应后繁琐的纯化操作。研究团队认为,借助该实用合成方案可常规制备enlicitide类似物,为药物性能优化与后续药物化学研究提供助力。总体而言,本研究中的所有合成操作与策略,为获取结构复杂的enlicitide及其类似物开辟了新前景,有助于研发高效的enlicitide类候选药物,以响应新型强效降脂药物的临床需求。
Cite this: Zhao, X.; Li, X.; Zhang, S.; Song, W.; Fang, H.; Jin, K. Efficient Synthesis of Enlicitide Chloride through Convergent Solution-Phase and Hybrid Solution–Solid-Phase Strategies. Precision Chemistry 2025. https://doi.org/10.1021/prechem.5c00107