英文原题:Tailoring ZnOx Species Confined on ZrO2 Support for Enhanced CO Hydrogenation
通讯作者:傅强,中国科学院大连化学物理研究所
作者:Le Lin (林乐), Xiaoyuan Sun (孙晓缘), Haoran Jia (贾浩然), Xiaohui Feng (冯小辉), Yingjie Wang (汪英杰), Rentao Mu (慕仁涛), Qiang Fu (傅强), Xinhe Bao (包信和)
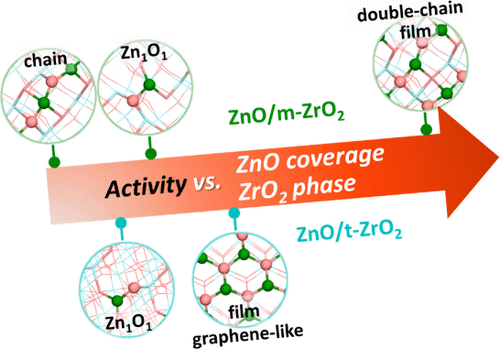
研究背景:
催化合成气转化到碳氢化合物,如低碳烯烃、芳烃和汽油,是一条重要的碳资源利用制高附加值化学品的方法。2016年,氧化物–分子筛(OXZEO)双功能催化概念被提出并得到了不断发展,其特点是将CO活化(在氧化物/OX表面)和C–C偶联(在分子筛/ZEO孔道)反应分离,一步法实现从合成气到低碳烯烃的高效转化。大量研究表明,复合氧化物在该体系中表现出优异的催化性能。ZnZrOx复合氧化物因其无毒性、高选择性等特点,有明显的研发潜力。然而,由于复合氧化物的结构复杂、可调维度多,导致其催化剂的精准设计仍充满挑战。
据文献报道,ZnZrOx催化剂以ZrO2为主体,只需微量的ZnO(如0.5%摩尔占比)即可发挥出显著的催化CO加氢活性,这些ZnOx物种多以单点或团簇状结构负载于ZrO2载体表面。就活性而言,有研究对比不同ZrO2晶相负载的ZnOx催化剂样品,发现单斜m-ZrO2上的ZnOx(ZnO/m-ZrO2)活性比四方相ZnO/t-ZrO2的更高。还有研究表明,对于界面催化剂,盖层ZnO的覆盖度、形貌等对催化性能有一定的调变作用。因此,利用多尺度理论模拟,对ZnZrOx复合氧化物界面体系进行全面的、原子尺度的探究意义重大,将有助于进一步实现高效催化剂的创新研制。
内容介绍:
我们基于两种实验上常用的ZrO2晶相,单斜(m-ZrO2)和四方(t-ZrO2)相,进行了表面态研究。每种晶相分别取三种优势的晶面,优化不同Zr/O化学计量比的表面结构并做从头算热力学分析,确定了实验制备条件到反应条件下的稳定表面态。从筛选出的ZrO2载体表面,继续构建不同ZnxOy化学计量比的ZnxOy/ZrO2界面模型,进行从头算热力学计算得到界面相图(图1)。发现m-ZrO2(11)(简称M(11))表面上最稳定的ZnOx物种是Zn4O4,其界面形成能比原始表面更正,说明该表面难以稳定ZnOx来形成热力学稳定的ZnO/M(11)界面;类似的,t-ZrO2的T(101)表面也难以稳定ZnOx。相比而言,M(001)、M(100)、T(100)和T(111)表面可分别稳定不同的ZnOx物种,形成热力学稳定的界面结构。特别地,M(001)上可形成三种ZnOx物种:双链状的Zn8O8单层膜、Zn4O4单链和Zn1O1单点结构,我们推测通过控制ZnO覆盖度实现这三种界面的可控制备。借助从头算分子动力学模拟,我们发现三种热力学稳定的ZnO/M(001)也表现出动力学稳定性,即在反应温度(673 K)下进行20 ps的动力学优化模拟,仍保持整体构型稳定,仅有局域的配位数变化。机理分析可知,M(001)与ZnO的界面结合能(Eadh)为−5.43 eV/Zn,比ZnO内聚能(Ecoh = −4.01 eV/Zn)还负,表明M(001)对ZnO有强的界面限域能力,而M(11)与ZnO的Eadh仅为−3.25 eV/Zn,表现出弱的界面作用,这本质上源于M(001)表面的Zr/O配位欠饱和。
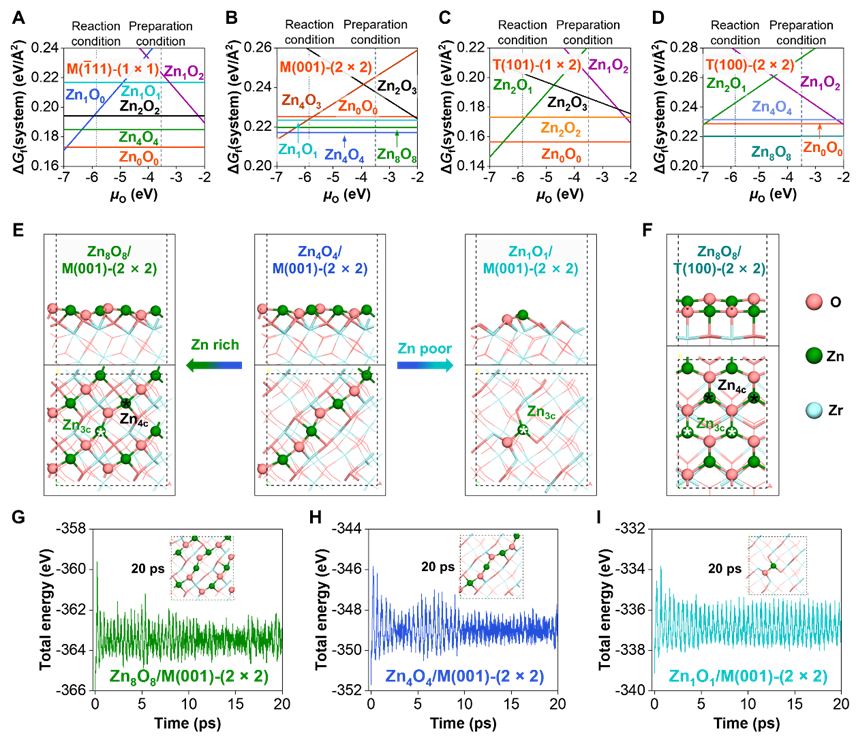
图1. 结合从头算热力学和分子动力学确定的ZnO/ZrO2界面结构及稳定性
接着对上述稳定或介稳的ZnO/ZrO2界面进行反应机理和活性研究(图2)。发现这些界面结构对CO和H2皆是弱吸附活化的,导致CO只能通过H助的方式进行活化,即连续加氢生成甲醇。微观动力学模拟表明,薄膜状的限域ZnOx物种具有比单链、单点状结构更高的本征活性,就ZrO2晶相而言,单斜相m-ZrO2上的ZnO薄膜比四方相t-ZrO2上的活性更高,其中Zn8O8/M(001)界面表现出最高的甲醇生成活性。进一步的电子和几何结构分析发现,CO和H2的活化和后续CHxO中间体的演变是多位点反应的,这要求活性中心必须兼具两方面的属性优势:(1)活泼Zn位点促中间体吸附活化;(2)多Zn位点协同稳定中间体或过渡态。有趣地是,在上述界面体系中,Zn8O8/M(001)恰好能提供活泼的、充足的Zn位点,实现中间体的稳定吸附和过渡态的能级降低,高效地催化CO加氢到甲醇。
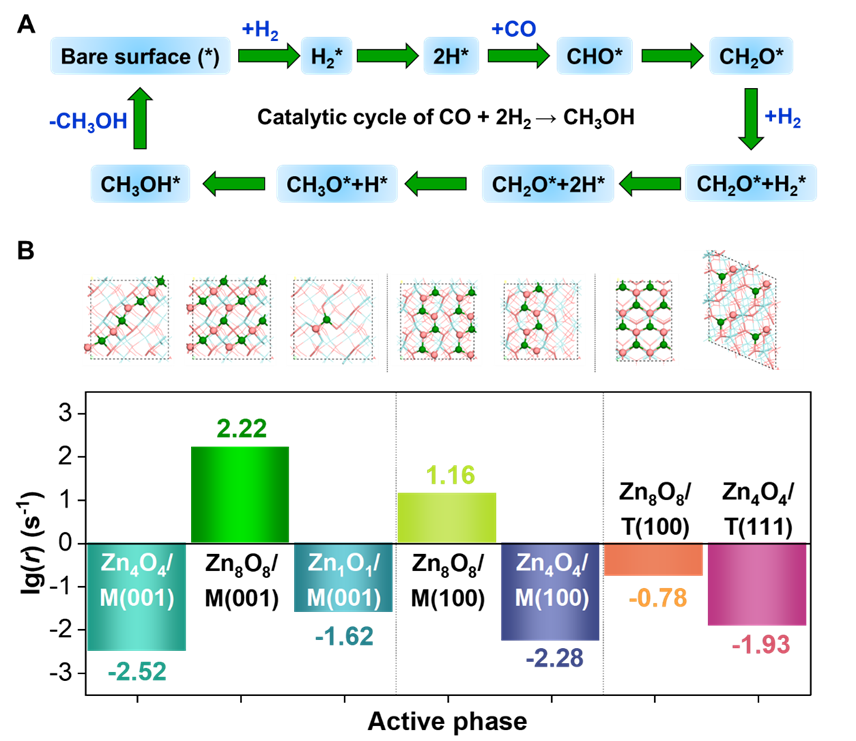
图2. 微观动力学模拟得到不同界面的甲醇生成活性对比
总结/展望:
我们从理论角度揭示了ZnO/ZrO2界面结构对ZrO2晶相和ZnO覆盖度的依赖关系。惰性的ZrO2表面(如单斜相的(11)面和四方相的(101)面)无法有效地限域住ZnOx物种,而活性表面(如M(001)、M(100)、T(100)和T(111))通过更强的界面粘结作用(更负的界面结合能Eadh)可稳定多种ZnOx结构。这些限域ZnOx物种对CO和H2吸附能力较弱,利于CO连续加氢生成甲醇,而不是直接或氢助CO解离。
就ZnO覆盖度而言,单层ZnO结构比单链或单点结构展现出更优的甲醇生成活性,这是因为单层结构有足够的位点和空间来稳定中间体和过渡态,使反应势能面更平坦。对比不同的ZrO2晶相,M(001)面限域的ZnOx薄膜(双链状)比T(100)面上的ZnOx薄膜(石墨烯状)有更高的甲醇生成活性,这源于两界面体系中的Zn位点差异:前者的八面体Znoct比后者的三角形Zntri有更高的d带中心,活性更高。这些活性位的配位环境与本征电子结构的差异,突显了ZnO覆盖度与ZrO2晶相在优化ZnZrOx复合氧化物催化剂中的关键作用,为进一步精准设计氧化物界面催化剂提供了思路。
Cite this: Lin, L.; Sun, X.; Jia, H.; Feng, X.; Wang, Y.; Mu, R.; Fu, Q.; Bao, X. Tailoring ZnOx Species Confined on ZrO2 Support for Enhanced CO Hydrogenation. Precision Chemistry 2025. https://doi.org/10.1021/prechem.5c00022.