英文原题:Pd-Catalyzed Stereospecific Glycosyl Cross-Coupling of Reversed Anomeric Stannanes for Modular Synthesis of Nonclassical C-Glycosides
通讯作者:朱峰,上海交通大学;年永,南京中医药大学;李英姿,西班牙加泰罗尼亚化学研究所
作者: Guoqiang Cheng (程国强), Bo Yang (杨波), Yang Han (韩阳), Wei Lin (林炜), Siyuan Tao (陶思远), Yong Nian (年永),* Yingzi Li (李英姿),* Maciej A. Walczak, and Feng Zhu (朱峰)*
研究背景:
芳基 C-糖苷 1广泛存在于各种具有生物活性的天然产物和药物化合物中,因其具有更稳定的 C−C 键而备受青睐。与具有水解不稳定 C−O 键的 O-糖苷相比,其在体内的代谢稳定性更高。非经典(Nc)芳基C-糖苷3是一类特殊的亚类,其特征是在吡喃糖的C-5位或呋喃糖的C-4位带有芳基取代基,而经典芳基C-糖苷则在端基C-1位带有芳基取代基。由于其独特的糖苷键连接方式,非经典芳基C-糖苷已被证明是非常有前景的抗癌剂、抗生素或糖尿病抑制剂。在某些情况下,其治疗效果甚至优于经典芳基C-糖苷。例如,经典芳基C-葡萄糖苷2达格列净是美国FDA批准上市的SGLT2(钠-葡萄糖协同转运蛋白2)抑制剂,而非经典芳基C-葡萄糖苷4索塔列净不仅是SGLT1和SGLT2的双重抑制剂,还对糖尿病并发的心血管疾病有治疗效果(图1A)。尽管非经典芳基C-糖苷具有非常吸引人的生物学特性,其合成却并未受到足够的关注。与经典C-糖苷合成的广泛研究相比,非经典C-糖苷的合成明显滞后且面临诸多挑战,阻碍了更多具有生物医药价值的糖类药物的开发。传统的非经典芳基C-糖苷合成方法包括ZnBr2介导的芳基锌试剂对4α-环氧吡喃糖的顺式选择性加成反应,以及芳香醛7和Danishefsky二烯8的选择性[4+2]环加成反应,生成糖烯衍生物9(图1B)。然而,这些策略面临原料获取困难、模块化程度差、底物范围有限、功能团耐受性差等障碍。
近期,自由基糖苷化反应在糖化学合成领域受到了广泛关注,成为合成经典C-糖苷的高效途径。此类合成方法具有多种优势,包括底物范围广、反应条件温和、操作简单以及官能团耐受性良好,从而推动了涉及非经典糖基自由基的糖基化反应研究,以制备非经典C-糖苷。例如,通过 N-杂芳烃与由糖醛酸或 α-烷氧基酰基碲化物产生的非经典糖基自由基的 Minisci 型 C−H 糖基化反应来合成非经典杂芳基 C-糖苷 11,已进行了大量研究,其中 Vismara、Inoue、Li 和 Liu 做出了重要贡献。这些巧妙的方法为目标化合物提供了简化的合成途径,并标志着显著的进步。然而,这些方法需要化学计量的氧化剂和自由基引发剂以确保反应顺利进行。因此,寻找能够高效精准合成非经典C-糖苷的策略仍是该领域的关键目标。
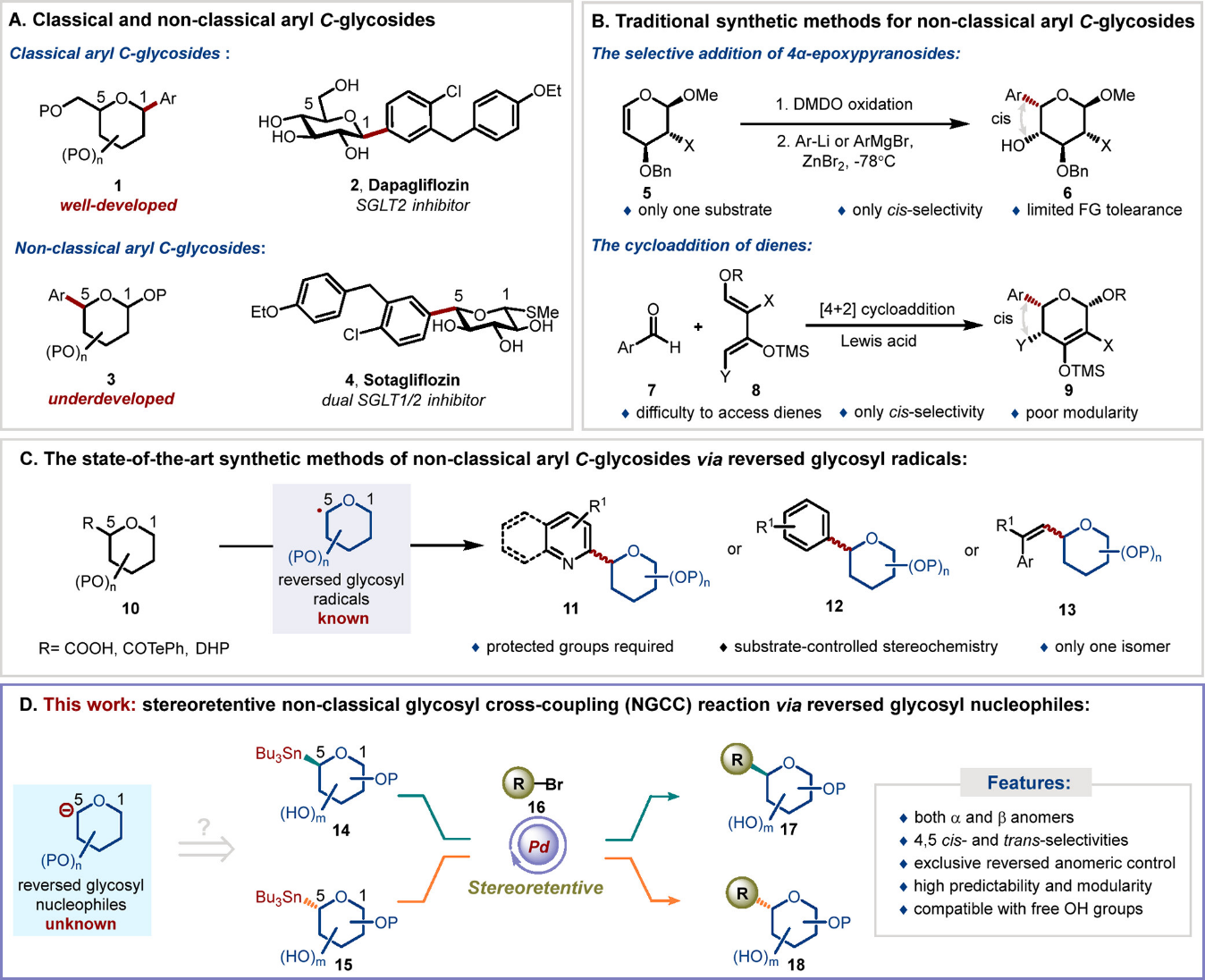
图1. 非经典 C-糖苷化研究:总体概述和作者的工作。
近年来,4-Nc-糖基-1,4-二氢吡啶(4-Nc-糖基DHP)试剂因其在合成非经典芳基C-糖苷中的潜力而备受关注(图 1C)。2018年,宾夕法尼亚大学Molander课题组报道了一种优雅的镍/光氧化还原双催化非经典糖基化反应,利用4-Nc-糖基DHP与(杂)芳基溴化物反应,得到非经典芳基C-糖苷12,其d.r.值从1:1.5到>20:1不等。值得注意的是,糖和芳香族骨架都与糖苷键构型密切相关,在某些情况下会产生底物控制的产物。尽管作者证明了通过逐一修改配体骨架可以提高非对映选择性,但非对映选择性与底物结构之间的关系仍然难以捉摸。南开大学的陈弓教授和何刚研究员团队随后报道了一种新颖的自由基方法,通过可见光照射下的4-Nc-糖基DHP与N-杂芳烃的Minisci型糖苷化,成功实现了非经典芳基C-糖苷的非对映选择性合成。值得注意的是,糖苷键的立体构型完全由糖骨架决定,不受杂芳基骨架的影响,从而表现出优异的立体选择性。最近,斯德哥尔摩大学的Olofsson课题组开发了一种高效的自由基介导C−C键形成新策略。该策略通过光氧化还原催化4-Nc-糖基DHP与乙烯基苯并索酮反应,生成非经典乙烯基C-糖苷13。众所周知,由于糖环的立体电子和空间效应可以显著影响糖基自由基的立体构型偏好,通过自由基糖基化方法同时获得α-和 β-糖基异构体一直具有挑战性。因此,迫切需要一种简洁、高度可控且可预测的非经典 C-糖基化策略来有效制备各种结构复杂的非经典 C-糖苷。
糖基金属亲核试剂参与的过渡金属催化糖基交叉偶联 (GCC) 反应代表了一种新颖的糖苷化反应设计策略。作者前期成功开发了由 Pd 催化的 α-和 β-经典糖基锡试剂与芳基卤化物的立体专一性交叉偶联反应,同时实现了α-和 β-经典芳基 C-糖苷的精确合成。该方法具有广泛的底物范围、出色的官能团兼容性和完美的糖苷键立体选择性。在此糖苷化反应中的关键立体控制步骤是立体构型保持的转金属化步骤,能够实现从原料到产物的糖苷键构型的手性转移,而完全不受糖和底物的立体和电子环境的影响。这一标准对于评估任何糖基化方法的潜在可扩展性至关重要。受到前期工作的鼓舞,作者旨在开发一种立体特异性非经典糖基交叉偶联策略(Non-classical Glycosyl Cross-Coupling, NGCC)。
内容介绍:
近日,上海交通大学朱峰副教授、南京中医药大学年永副教授和西班牙加泰罗尼亚化学研究所李英姿博士等设计开发了一种Pd催化非经典糖基锡试剂与芳基和烯基亲电试剂参与的立体专一性C-糖苷化反应,实现了非经典C-糖苷的高度模块化合成。
作者期望开发的非经典C-糖化反应具有以下特点(图 1D):1)兼容各种常见的非经典α-和β-糖基锡试剂;2)适用于构建4,5-顺式和反式的糖苷键;3)完全的立体构型保持:能够实现非经典糖基锡试剂的立体构型保持的转金属化,从而高效、精确地构建非经典C-糖苷键;4)具有高度的可控性、可预测性、立体选择性和化学选择性;5)兼容无保护的糖基底物。为了成功开发该方法,需要解决以下关键问题:1)建立种类丰富的非经典糖基锡试剂库;2)解决非经典和经典糖基锡烷之间的结构差异可能带来的反应活性的影响,并筛选新的C−C键形成条件;3)突破经典C-糖苷化反应中sp2碳亲电试剂的局限性,实现烯基C-糖苷的合成。
针对上述挑战,作者首先设计并合成了一系列非经典糖基锡试剂(图2)。这些结构多样且光学纯的非经典糖基锡试剂为开发通用、高度模块化、立体选择性的非经典糖苷化反应奠定了基础。如方案 1 所示,通过对 4-脱氧戊烯苷 (4-DPs) 19进行一系列合成转化,获得了两种糖基异构体锡试剂(21a-21h)。受到ZnBr2介导的4-环氧吡喃苷顺式加成的启发,作者还通过与Bu3SnZnBr的顺式开环反应成功获得了4,5-顺式-α-锡烷22a。此外,通过将单糖锡试剂转化为寡糖(24a、24b),成功拓展了非经典糖基锡试剂的多样性和复杂性。
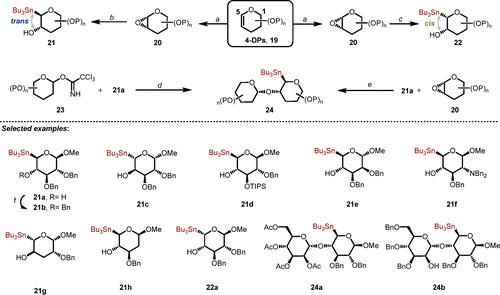
反应试剂和条件: (a) Oxone® (4.0 equiv), acetone, NaHCO3, CH2Cl2/H2O, 0 oC to rt, 99%; (b) n-Bu3SnMgMe (3.00 equiv), THF, - 20 oC, 12%-41%; (c) i. n-Bu3SnLi (6.00 equiv), ZnBr2 (7.00 equiv), THF, -78 oC to -30 oC, 16 h, 10%; (d) TMSOTf (0.25 equiv), 5Å MS, THF, -20 oC, 30%; (e) Ph3PAuCl (0.30 equiv), AgOTf (0.30 equiv), 4Å MS, -78 oC to rt, 3 d, 33%; (f) BnBr (2.0 equiv), KHMDS (1.5 equiv), THF, 0 oC to rt, 2.5 h, 93%.
图2. 合成非经典端基锡烷
在获得一系列非经典糖基锡试剂后,下一步是筛选最佳的催化条件以实现立体专一性的非经典糖基交叉偶联 (NGCC) 反应(表 1)。作者首先使用标准的 Pd2(dba)3/Jackiephos L1 催化系统,该系统因成功合成经典芳基C-糖苷而知名,用于研究非经典异头锡烷 21b和 4-溴联苯16a的模型反应。经过详细的条件优化,最终以97%的NMR收率和93%的分离收率获得目标化合物(entry 15)。令人惊讶的是,这种Pd催化的非经典糖基锡试剂参与的交叉偶联反应可以在 1,4-二氧六环和水1:1 的混合溶剂中成功进行,并以 74% 的收率生成 25a(entry 18)。这表明该方法有望实现水溶性生物大分子的后期糖多样性修饰。
表 1. 反应条件优化a
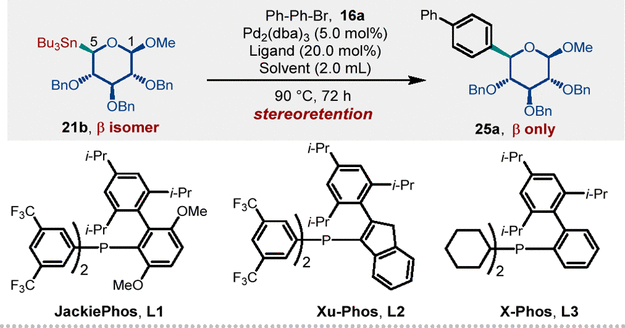
Entry | Ligand | Additives | Solvent | NMR Yield |
1 | L1 | Ag2CO3 | 1,4-Dioxane | 77% |
2 | L1 | Ag2CO3 | Toluene | 59% |
3 | L1 | Ag2CO3 | DMF | 54% |
4 | L1 | Ag2CO3 | tBuOH | 79% |
5 | L1 | Ag2CO3 | 1,4-Dioxane/tBuOH 1:1 | 81% |
6b | L1 | Ag2CO3 | 1,4-Dioxane/tBuOH 1:1 | 82% |
7b | L2 | Ag2CO3 | 1,4-Dioxane/tBuOH 1:1 | 54% |
8b | L3 | Ag2CO3 | 1,4-Dioxane/tBuOH 1:1 | 17% |
9b,c | L1 | Ag2CO3 | 1,4-Dioxane/tBuOH 1:1 | 50% |
10b,d | L1 | Ag2CO3 | 1,4-Dioxane/tBuOH 1:1 | 11% |
11b | L1 | Ag2O | 1,4-Dioxane/tBuOH 1:1 | 83% |
12b | L1 | AgF | 1,4-Dioxane/tBuOH 1:1 | 86% |
13b,e | L1 | AgF | 1,4-Dioxane/tBuOH 1:1 | 72% |
14b,f | L1 | AgF | 1,4-Dioxane/tBuOH 1:1 | 91% |
15b,f,g | L1 | AgF | 1,4-Dioxane/tBuOH 1:1 | 97% (93%) |
16b,h | L1 | AgF | 1,4-Dioxane/tBuOH 1:1 | N.R. |
17b,f,g,i | L1 | AgF | 1,4-Dioxane/tBuOH 1:1 | 8%, 93%,16% |
18 | L1 | AgF | 1,4-Dioxane/H2O 1:1 | 74% |
a标准反应条件: 16a (0.10 mmol), 21b (0.20 mmol), Pd2(dba)3 (5 mol%), ligand (20 mol%), CuCl (1.0 equiv), Agsalts (1.0 - 2.0 equiv), 1,4-dioxane (2.00 mL), 90 °C, 72 h, N2, isolated yields. b48 h was used. cCuBr was used. dCuI was used. e110 °C was used. f70 °C was used. gPd2(dba)3 (2.5 mol%) and ligand (10 mol%) were used. hWithout Pd2(dba)3 or Cu salts. i4-Chlorobiphenyl, 4-Iodobiphenyl, or trifluoromethanesulfonic acid 4-biphenylyl ester was used. The stereochemical outcome was determined by 1H NMR analysis of crude reaction mixtures.
在确定最佳反应条件后,作者开始研究亲电偶联试剂的范围。如图 3 所示,多种(杂)芳基溴化物在反应中表现出优异的兼容性,可以良好至优异的产率、高度立体专一性地获得相应的非经典芳基糖苷。值得一提的是,NGCC 反应不仅适用于芳基亲电试剂,还涵盖了苯并呋喃、苯并恶唑和吲哚等杂芳烃,它们也能够顺利进行非经典C-糖基化反应(25n-25p)。
非经典烯基C-糖苷是一类具有重要合成潜力的化合物,其合成面临巨大的挑战。目前仅有少数几种有效的方法可用于其合成。作者研究发现,Pd催化的非经典糖基锡试剂与烯基溴的交叉偶联反应在构建烯基C−C键时表现出优异的糖苷键构型控制和Z/E-构型保持(25q-25u')。该方法代表了首例能够同时实现非经典芳基和烯基 C-糖苷的立体选择性合成方法。
为了进一步探索非经典糖基交叉偶联方法的适用范围和多功能性,作者使用21b作为糖基供体,在标准反应条件下对多种市售药物和生物活性分子进行了后期糖基多样化。例如,氨基酸和二肽在标准条件下顺利反应,产率达到85-90%(25v-25x)。类似地,几种用芳基亲电试剂预先功能化的市售药物和小分子天然产物(25y-25ae),在标准条件下也能与非经典异头锡烷21b顺利偶联,产率超过91%。这些结果证明了该方法在合成可能具有生物活性的多种糖缀合物方面的广泛适用性和多功能性。
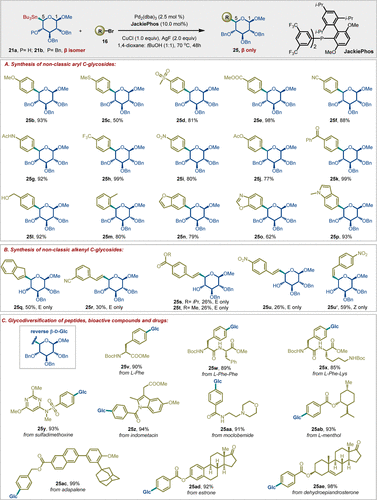
图3. 亲电偶联试剂的底物范围
作者进一步评估了非经典糖基锡试剂的底物范围,使用 4-溴苯甲酸甲酯作为模型底物。如图 4 所示,多种具有不同糖型的非经典糖基锡试剂在反应中表现出优异的反应性,以良好至优异的产率和完全的β-或α-选择性生成所需的偶联产物(27a-27f)。此外,4,5-顺式锡烷异构体(27g)也展现出完美的立体构型保持性。非经典芳基 C-葡萄糖胺27h也能以优异的产率获得。带有常见保护基如OTIPS、ONap和OAc的非经典端基锡烷在标准条件下表现出良好的兼容性(27i、27j和27m) 。无保护糖的立体选择性糖基化代表了一个极具挑战性的研究领域。在标准反应条件下,含有游离羟基的非经典糖基锡试剂表现出优异的反应性和化学和立体选择性(27a-27i、27k-27m)。值得注意的是,从糖基化反应中获得的寡糖锡烷24a和24b(如图1所示),无需修改标准条件即可成功应用于非经典糖基交叉偶联反应,生成含有(1→4)糖苷键的非经典C-二糖。
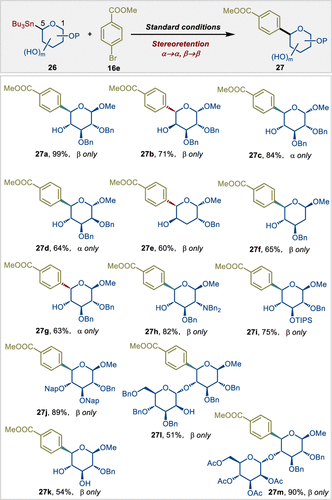
图4. 非经典糖基锡试剂的底物范围
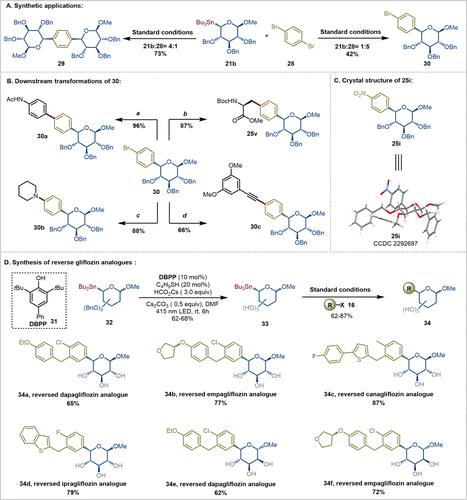
图 5. 合成应用和下游转化
为了验证该反应的实用性,作者进行了一系列合成应用和下游转化。在图 5A中,1,4-二溴苯与21b反应生成了双糖基化产物29,且具有完全的β-选择性。进一步优化后,作者发现通过调整亲电试剂的当量,可以轻松合成单糖基化产物 30,其中溴原子留在芳环上,为进一步的转化提供了反应手柄。在后续的经典交叉偶联转化中,如Suzuki反应、Sonogashira反应、C-N偶联和Stille偶联,都可以通过30参与的Pd催化交叉偶联反应顺利进行(图5B)。所有反应都表现良好,非经典位的糖苷键选择性均未受到影响。此外,通过X射线晶体学分析,明确了25i的立体化学(图5C)。
无保护的糖基供体与各种芳基卤化物的后期 C-糖苷化反应是快速构建 C-糖苷候选药物库的理想方法。然而,这一过程中如何控制糖苷键的立体选择性和克服多羟基带来的化学选择性挑战仍然是难题。为了应对这些挑战,作者的目标是展示当前方法在无保护糖苷化反应中的潜在应用。为避免使用苛刻的Birch还原条件,作者首先需要开发一种简便且高效的合成无保护非经典糖基锡试剂的方法。受到云南大学夏成峰教授团队开发的光化学方法高效脱除苄基保护基的启发,作者成功将这一光催化苄基脱除方法应用于非经典糖基锡试剂的脱苄基,从而获得了无保护的非经典糖基锡试剂33。随后,作者将这种无保护的非经典糖基锡试剂33与一系列SGLT2抑制剂的芳基亲电试剂进行偶联,生成了相应的非经典gliflozin药物类似物。这些类似物有望成为新的抗糖尿病药物(图5D)。
非经典C-糖基化修饰为药物分子的糖基化开辟了新的方向,有望显著提高其生物活性。为验证这一假设,作者与南京中医药大学年永副教授合作,设计并合成了非经典甘露糖基吲哚美辛35(图6A)。研究结果显示,相比于母体化合物吲哚美辛36,具有C-5-糖基结构的化合物35在小鼠肝微粒体中展现了增强的代谢稳定性和更高的动力学溶解度(图6B)。
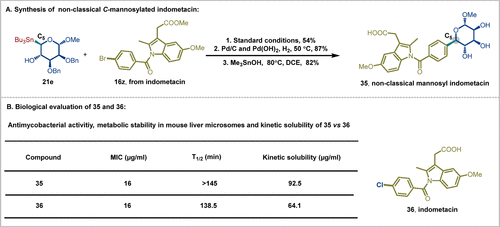
图6. 生物学评估
在评估非经典糖基锡试剂的适用范围时,作者发现C1位的OMe基团对反应的立体特异性没有显著影响,但却会影响反应效率。为深入探究非经典糖基锡试剂的空间位阻与反应活性之间的关系,作者进行了控制实验(图7)。有趣的是,尽管21b的空间位阻大于21k,其偶联产率却更高。这表明,尽管21k中的OMe基团在理论上可能引入有利的空间位阻,但实际上对转移金属化的影响却是不利的。此外,相比于21b和21k,21i的活性较低,说明糖的类型对反应效率的影响要大于C1-OMe基团的立体构型。值得注意的是,在添加剂KF或AgF的存在下,模型反应分别以69%和97%的产率生成了目标产物25a。这引发了关于AgF和KF在反应机制中作用的重要问题。
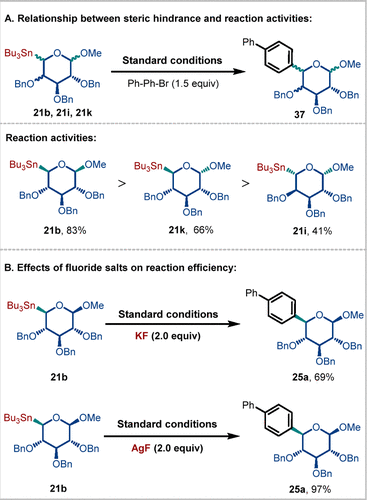
图7. 控制实验
为了探究非经典糖基锡试剂与经典糖基锡试剂反应性差异的根本原因,作者与西班牙加泰罗尼亚化学研究所李英姿博士合作,利用密度泛函理论(DFT)对该反应进行了计算分析,如图8所示。研究表明,该交叉偶联反应的启动阶段由Pd(0)物种与溴苯的氧化加成开始,形成苯基钯(II)溴化物复合物(int2)。接着,非经典异头锡烷21b’通过四元环过渡态(ts5)进行立体保持的转金属化,此步骤的活化能高达39.0 kcal/mol。有趣的是,研究发现AgF的存在可以促进这一转金属化过程。AgF的配位通过形成六元环过渡态(ts4)显著降低了转金属化的能垒至21.5 kcal/mol,形成中间体int4。随后,int4通过无能垒的快速还原消除生成目标产物25a’,并再生PdL催化剂。此外,作者还考虑了一种替代路径,即在int2中进行AgF辅助的卤化物交换,以生成更稳定的Pd (II)氟化物物种int3,然后进行转金属化(ts2)。然而,该路径的活化能为34.5 kcal/mol,因此不利于反应。研究还探讨了在int3中芳基翻转形成int3'的可能性,预计这一过程会显著降低转金属化的能垒(ts2',ΔG‡ = 21.8 kcal/mol)。然而,int3难以通过P-Ar键旋转异构化为int3'(ΔE‡ = 28.9 kcal/mol),因此这一途径被排除。
作者随后进行了计算分析,以深入了解不同构型的非经典糖基锡试剂之间的反应性差异,如图8所示。转移金属化过程对总反应速率至关重要,且为速率决定步骤。在将底物 21b'替换为 21k' 后,过渡态 (ts4_21k') 的活化能增加到 26.7 kcal/mol,这主要是由于ts4_21k'中向下取向的 OMe 基团与配体的芳基官能团之间的空间位阻。相对而言,底物21i'无法形成有利的六元环转金属化过渡态,而是形成了活化能为30.2 kcal/mol的四元环过渡态(ts2_21i')。计算结果表明,底物21i'的活性显著低于21b'和21k'。作者进一步分析了AgF和KF对反应的影响。初步比较显示过渡态之间的反应能量差异不显著。作者深入研究了AgF和KF二聚体在反应中的稳定性。AgF二聚体的过渡态能垒为22.4 kcal/mol,而KF二聚体的能垒则显著更高,为34.2 kcal/mol。显著的能垒差异表明这两种二聚体在反应性上存在显著不同。此外,AgF和KF二聚体在转化为单体的能量分别为23.8 kcal/mol和28.2 kcal/mol,这表明AgF的优越性能可能与其较低的能垒和更有利的反应路径有关。
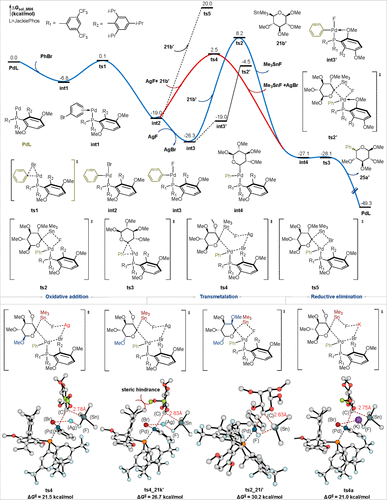
图8. 非经典糖基锡试剂与溴苯进行糖基交叉偶联反应的能量分布图
总结/展望:
朱峰副教授、年永副教授和李英姿博士等开发了一种简洁而通用的糖类合成方法,通过 Pd 催化非经典糖基锡试剂参与的糖基交叉偶联反应来精准合成非经典 C-糖苷。该方法巧妙地利用立体构型保持的转金属化步骤精确地控制糖苷键的构型,而完全不受糖和底物的立体和化学环境的影响,从而区别于现有的非经典 C-糖基化方法。该方法展现了高度的可控性、可预测性和模块化,能够兼容广泛的底物,具有优异的官能团耐受性,并始终保持高化学选择性和立体特异性。立体专一性的非经典C-糖基化的开发既及时又重要,它填补了立体专一性合成非经典C-糖苷的研究空白,并促进了这些化合物作为潜在糖类药物的广泛生物学研究。因此,这一策略不仅为药物相关的非经典C-糖苷合成提供了有力工具,还推动了糖基金属亲核试剂参与的糖基交叉偶联反应(Glycosyl Cross-Coupling ,GCC)的发展。
Cite this: Cheng, G.; Yang, B.; Han, Y.; Lin, W.; Tao, S.; Nian, Y.; Li, Y.; Walczak, M. A.; Zhu, F. Pd-Catalyzed Stereospecific Glycosyl Cross-Coupling of Reversed Anomeric Stannanes for Modular Synthesis of Nonclassical C-Glycosides. Precision Chemistry 2024, 2 (11), 587–599. https://doi.org/10.1021/prechem.4c00042.