中国科学技术大学蒋彬教授课题组在分子与金属表面碰撞的非绝热动力学模拟方面取得重要进展。研究成果以“包含振动-电子耦合的分子在金属表面的第一性原理非绝热动力学(First-principles Nonadiabatic Dynamics of Molecules at Metal Surfaces with Vibrationally Coupled Electron Transfer)”为题,于2024年7月19日发表在《物理评论快报》(Physical Review Letters)上(Phys. Rev. Lett., 2024, 133, 036203)。
由于金属表面连续的电子能级,分子与金属表面相互作用时,金属表面的电子很容易被激发,导致分子与金属表面间的非绝热能量转移。包括分子在金属表面极短的振动态寿命、化学电流、氢原子从金属表面散射后剧烈的能量损失、以及振动激发态的NO和CO分子从金属表面散射后的振动弛豫在内的众多实验现象都证明非绝热能量转移广泛存在于各种界面过程中,因此研究非绝热能量转移对于理解化学吸附、电化学、等离激元催化等界面过程具有重要意义。然而,分子与金属表面相互作用的过程中,分子振动、转动、平动与表面声子和电子会耦合在一起,导致极为复杂的能量转移过程,因此,准确描述涉及电子转移的分子在金属表面的非绝热动力学是理论界长期面临的挑战。
为了解决这一问题,研究团队提出了“约束密度泛函理论+嵌入原子神经网络+独立电子面跳跃”的模拟策略,并将该策略用于CO分子从Au(111)表面散射过程中的能量转移动力学模拟。如图1所示,研究人员首先用约束密度泛函理论计算了众多构型的CO分子在金属表面的电子转移透热态,并用嵌入原子神经方法拟合相应的全维势能面,最后用独立电子面跳跃方法模拟分子散射过程中的能量转移过程。研究结果显示,独立电子面跳跃模拟得到的高振动态CO(vi=17)分子散射后的振动末态分布与实验很接近(图2),低振动态CO(vi=2)散射后的振动弛豫概率、平均平动能以及散射角分布也都被理论模拟比较好地重现了(图3)。特别值得一提的是,模拟结果还揭示了不同初始振动态下不同的能量传递通道:在高初始振动态下,分子振动能主要传递到表面电子和分子平动,而在低初始振动态下,分子振动能则只传向表面电子。这一系列的发现对于理解分子-表面体系的能量传递过程有着重要的意义。此外,这套模拟策略有望用于研究一些复杂过程的能量转移动力学,比如光/电化学和等离激元催化过程。
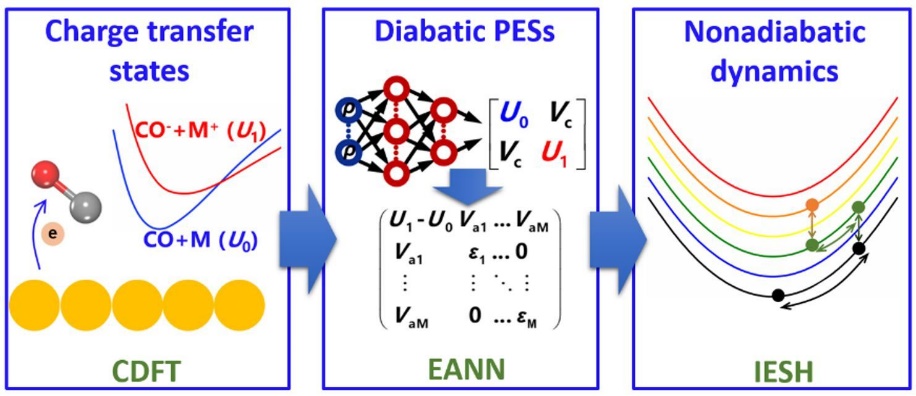
图1 分子在金属表面非绝热动力学模拟的工作流程示意图
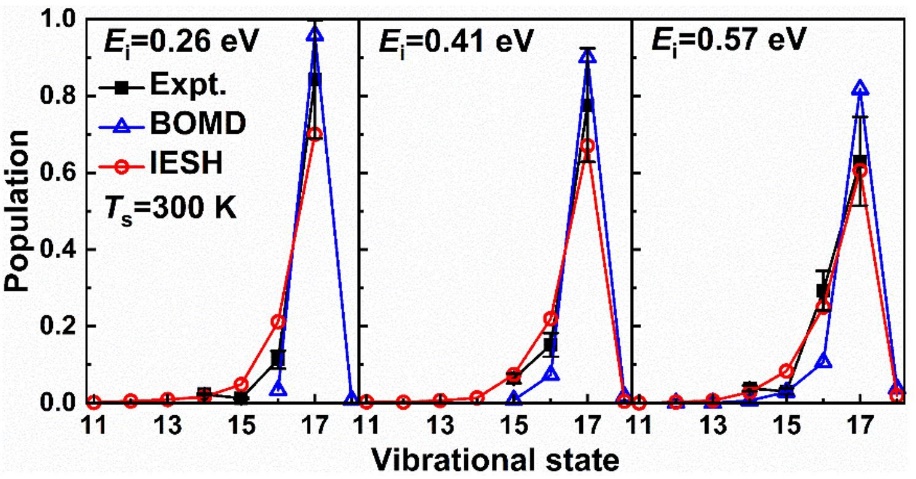
图2 CO(vi=17)分子从Au(111)表面散射后的振动末态分布
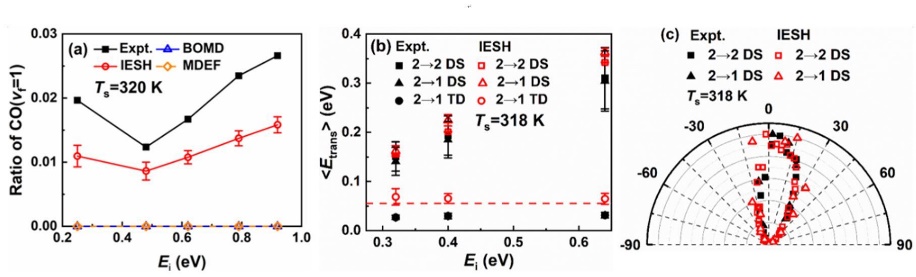
图3 CO(vi=2)分子从Au(111)表面散射后的一些动力学性质
中国科学技术大学化学物理系博士生孟刚为该论文的第一作者,University of Warwick的Reinhard J. Maurer教授与中国科学技术大学的蒋彬教授为共同通讯作者,西湖大学的窦文杰教授合作参与了研究。该工作得到了中国科学院战略先导科技专项、量子科学与技术创新项目、中国科学院稳定支持基础研究领域青年团队、基金委创新群体、重点项目等基金的资助。计算模拟工作在中国科学技术大学超级计算中心、合肥先进超算中心等完成。
论文链接:https://link.aps.org/doi/10.1103/PhysRevLett.133.036203
(化学与材料科学学院、精准智能化学重点实验室、科研部)